

Cambiando el futuro con 6 gotitas de sangre
Cuando se convierte en madre, es normal tener muchas preguntas sobre el cuidado de su bebé y su salud. Una de las pruebas que se realizará a su bebé poco después del nacimiento es el Tamiz Metabólico Neonatal.
¿Qué es el Tamiz Metabólico Neonatal?
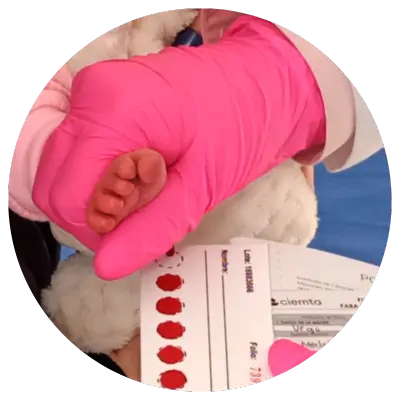
Es una prueba que se realiza a los recién nacidos para la detección oportuna de alguna enfermedad o deficiencia metabólica antes de que se manifieste, para dar tratamiento adecuado y oportuno, limitando complicaciones graves como discapacidad intelectual, daño cerebral, retraso en el crecimiento, problemas de desarrollo, y en algunos casos la muerte.
¿Por qué es importante realizarlo a mi bebé?
Porque puede detectar trastornos metabólicos de manera temprana, lo que permite iniciar un tratamiento inmediato y reducir el riesgo de complicaciones a largo plazo. La detección temprana y el tratamiento adecuado también pueden prevenir discapacidades cognitivas y físicas, así como la muerte prematura.
¿Cuándo y cómo se realiza?
La prueba consiste en tomar unas gotas de sangre del talón de tu bebé, lo ideal es realizarlo al segundo y quinto día de nacido, pero se puede hacer hasta los 30 días.
La muestra se coloca en un papel filtro especial que se envía al laboratorio especializado para su análisis; como lo es ciemta. Ahí se evalúa y se busca niveles elevados de ciertas sustancias en la sangre que podría indicar la presencia de una enfermedad metabólica.
El resultado se obtiene después de unos días y es entregado a los padres o tutores del bebé. No te asustes, la prueba es más sencilla de lo que crees, conoce en el siguiente video cómo se realiza.
¿Qué enfermedades detectamos?
Trastornos metabólicos:
Los trastornos metabólicos hereditarios se clasifican en distintos grupos. Se agrupan en función de la sustancia afectada y de si se acumula en exceso porque no puede descomponerse o es demasiado baja o falta.
Trastornos por deficiencia enzimática
Son trastornos genéticos (hereditarios) poco comunes por los cuales el cuerpo es incapaz de convertir los alimentos en energía de manera apropiada. Dichos trastornos generalmente son causados por defectos en proteínas específicas (enzimas) que ayudan a descomponer (metabolizar) partes del alimento.
Deficiencia de biotinidasa (BTD)
La deficiencia de biotinidasa se produce cuando una enzima específica (un tipo de proteína) en el cuerpo, llamada biotinidasa, no puede liberar biotina (una de las vitaminas B) de los alimentos que comemos, por lo que puede usarse para energía y crecimiento.
Acidemias orgánicas
Son un grupo de enfermedades causadas por un bloqueo enzimático específico del catabolismo de los aminoácidos de cadena ramificada.
Deficiencia de glucosa 6 fosfato deshidrogenasa (G6PD)
Es una anemia hemolítica constitucional poco frecuente debida a un trastorno enzimático caracterizado por deficiencia grave de glucosa-6-fosfato deshidrogenasa (típicamente <10% de actividad enzimática residual) asociada a anemia hemolítica crónica no esferocítica de gravedad muy variable.
Los bebés corren el riesgo de desarrollar ictericia neonatal (que puede conducir a querníctero), cálculos biliares, reticulocitosis y esplenomegalia, y presentan una mayor susceptibilidad a los agentes oxidantes, que desencadenan episodios de hemólisis aguda.
Trastornos de la oxidación de los ácidos grasos
Son trastornos del metabolismo de los lípidos que están causados por una carencia o deficiencia de las enzimas necesarias para descomponer las grasas, dando lugar a retraso en el desarrollo mental y físico. Los trastornos de oxidación de los ácidos grasos se producen cuando los padres transmiten el gen defectuoso que causa estos trastornos a sus hijos.
Anemia de células falciformes (anemia drepanocítica o drepanocitosis)
Es una anomalía genética hereditaria de la hemoglobina (la proteína presente en los glóbulos rojos que transporta el oxígeno) caracterizada por la presencia de glóbulos rojos en forma de hoz (media luna) y anemia crónica, causada por una excesiva destrucción de dichos glóbulos rojos anómalos.
Trastornos lisosomales
Los lisosomas, la unidad de reciclaje de una célula, son sacos que contienen enzimas que descomponen componentes como azúcares y grasas en sustancias que el cuerpo puede usar. Cuando los lisosomas no funcionan correctamente, se denomina trastorno lisosomal (LD). Las personas con trastornos lisosomales no pueden descomponer los azúcares y las grasas, lo que hace que se acumulen en la célula. Esta acumulación tóxica puede causar problemas de salud. Los trastornos lisosomales pueden afectar el cerebro, el hígado, el bazo, los huesos, los músculos o el corazón.
Fabry
Es una enfermedad lisosomal multisistémica poco frecuente de origen genético caracterizada por manifestaciones cutáneas específicas (angioqueratoma), neurológicas (dolor), renales (proteinuria, insuficiencia renal crónica), cardiovasculares (miocardiopatía, arritmia), cocleovestibulares y cerebrovasculares (ataques isquémicos transitorios, accidentes cerebrovasculares).
Mucopolisacaridosis
Son un tipo de enfermedades de depósito lisosomal en las que las moléculas complejas de azúcar no se degradan normalmente y se acumulan en cantidades perjudiciales en los tejidos del organismo. El resultado es una apariencia facial característica y anomalías en los huesos, los ojos, el hígado y el bazo, a veces acompañadas de retraso mental.
Las mucopolisacaridosis ocurren cuando los progenitores transmiten el gen defectuoso que causa estos trastornos a sus hijos.
Gaucher (EG)
Es una enfermedad de depósito lisosomal que comprende 3 tipos principales (tipos 1, 2 y 3), una forma fetal y una variante con afectación cardiovascular (enfermedad de Gaucher-oftalmoplegia-calcificación cardiovascular o enfermedad similar a Gaucher).
Desórdenes endócrinos
Los trastornos endócrinos ocurren cuando hay un problema con la cantidad de hormonas (mensajeros químicos) en nuestro cuerpo. Los trastornos endócrinos identificados con el tamiz neonatal son la hiperplasia suprarrenal congénita (CAH) y el hipotiroidismo congénito (CH).
Hiperplasia suprarrenal congénita
Los bebés que nacen con hiperplasia suprarrenal congénita (CAH, por sus siglas en inglés) no pueden producir suficiente hormona cortisol, que ayuda a controlar la energía, los niveles de azúcar, la presión arterial y la forma en que el cuerpo responde al estrés de una lesión o enfermedad. CAH también puede afectar el desarrollo de los genitales y las hormonas de la pubertad.
Hipotiroidismo congénito
Esta es una condición en la que el bebé nace con muy poca hormona tiroidea. Los niveles bajos de hormona tiroidea sin tratar pueden provocar problemas de desarrollo mental y un crecimiento deficiente.
Trastornos de aminoácidos
Los trastornos del metabolismo de los aminoácidos son trastornos metabólicos hereditarios. Los aminoácidos, son los ladrillos que forman las proteínas, cumplen diversas funciones en el organismo. El cuerpo sintetiza algunos de los aminoácidos que necesita y obtiene otros de los alimentos. Los trastornos hereditarios del metabolismo de los aminoácidos son el resultado de cualquier defecto en la asimilación de los aminoácidos o en la capacidad del organismo para llevar los aminoácidos a las células.
Enfermedad de la orina de jarabe de arce
Este es un trastorno hereditario que es muy común en la población menonita. El trastorno es causado por la incapacidad del cuerpo para procesar adecuadamente ciertas partes de la proteína llamadas aminoácidos.
El nombre proviene del olor característico del jarabe de arce en la orina del bebé causado por el metabolismo anormal de las proteínas. Si no se trata, es potencialmente mortal ya en las dos primeras semanas de vida. Incluso con tratamiento, pueden ocurrir discapacidad severa y parálisis.
Fenilcetonuria (PKU)
Es un trastorno del metabolismo de los aminoácidos que se produce en bebés nacidos sin la capacidad de descomponer normalmente un aminoácido denominado fenilalanina. La fenilalanina, tóxica para el cerebro, se acumula en la sangre.
La fenilcetonuria ocurre cuando los progenitores transmiten a sus hijos el gen defectuoso que causa este trastorno.
Tirosinemia
Es un trastorno del metabolismo de los aminoácidos que está causado por la falta de la enzima necesaria para metabolizar la tirosina. La forma más frecuente de tirosinemia afecta sobre todo el hígado y los riñones. La tirosinemia ocurre cuando los padres transmiten a sus hijos un gen defectuoso que causa este trastorno.
Otros trastornos genéticos
Una enfermedad genética es una afección que se produce por alteraciones en los genes o cromosomas de las células del cuerpo. Estas alteraciones, llamadas mutaciones, pueden ser heredadas de los padres o surgir de forma espontánea. Las enfermedades genéticas pueden ser causadas por: Cambios en un único gen, Cambios en muchos genes, Daños en los cromosomas.
Inmunodeficiencia
combinada severa (SCID)
Afecta la forma en que el cuerpo combate las infecciones. Una persona con SCID no tiene un sistema inmunológico que funcione correctamente. Sin un sistema inmunitario que funcione, el cuerpo no puede combatir las infecciones.
Fibrosis quística (FQ)
Es un trastorno que afecta la respiración y la digestión (descomposición de los alimentos). Una persona con FQ produce una mucosidad espesa y pegajosa que bloquea las vías respiratorias de los pulmones, lo que dificulta la respiración. Esta mucosidad también puede dificultar que el cuerpo descomponga los alimentos.
Atrofia muscular espinal (AME)
Es un trastorno genético que afecta el control del movimiento muscular. LA AME es una enfermedad progresiva, lo que significa que empeora con el tiempo. Los niños con AME pierden la capacidad de caminar, comer y finalmente, respirar. Sin tratamiento, LA AME puede provocar la muerte en la primera infancia.
En ciemta contamos con 3 tipos de Tamiz Metabólico
Tamiz Metabólico Básico -16 enfermedades
HIPERPLASIA SUPRARENAL CONGÉNITA VIRALIZANTE SIMPLE
- HIPERPLASIA SUPRARENAL CONGÉNITA PERDEDORA DE SAL
- HIPOTIROIDISMO CONGÉNITO
- HIPERTIROTROPINEMIA
- DEFICIENCIA DE BIOTINIDASA
- FIBROSIS QUÍSTICA
- DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
- GALACTOSEMIA TIPO DUARTE
- GALACTOSEMIA CLÁSICA
- GALACTOSEMIA TRANSITORIA
- DEFICIENCIA DE GALACTOQUINASA
- DEFICIENCIA DE GALACTOSA EPIMERASA
- FENILCETONURIA
- HIPERFENILALANINEMIA (H-PHE)
- DEFECTO EN LA SÍNTESIS DEL COFACTOR DE BIOPTERINA (BIOP BS)
- DEFECTO DE REGENERACIÓN DEL COFACTOR BIOPTERINA (BIOP REG)
Tamiz Metabólico Ampliado -96 enfermedades
- HIPERPLASIA SUPRARENAL CONGÉNITA VIRALIZANTE SIMPLE
- HIPERPLASIA SUPRARENAL CONGÉNITA PERDEDORA DE SAL
- HIPOTIROIDISMO CONGÉNITO
- HIPERTIROTROPINEMIA
- DEFICIENCIA DE BIOTINIDASA
- FIBROSIS QUÍSTICA
- DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
- GALACTOSEMIA TIPO DUARTE
- GALACTOSEMIA CLÁSICA
- GALACTOSEMIA TRANSITORIA
- DEFICIENCIA DE GALACTOQUINASA
- DEFICIENCIA DE GALACTOSA EPIMERASA
- ENFERMEDAD DE HEMOGLOBINA S
- ENFERMEDAD DE HEMOGLOBINA C
- ENFERMEDAD DE HEMOGLOBINA S/C
- ENFERMEDAD DE HEMOGLOBINA E
- ENFERMEDAD DE HEMOGLOBINA D
- ENFERMEDAD DE CÉLULAS FALCIFORMES CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA C CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA E CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA H
- ENFERMEDAD DE HEMOGLOBINA S CON RASGO DE ALFA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA S/C CON RASGO DE ALFA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA G FILADELFIA
- ENFERMEDAD DE HEMOGLOBINA G CON RASGO DE ALFA TALASEMIA
- BETA TALASEMIA MAYOR
- ANEMIA DREPANOCÍTICA
- DEFICIENCIA DE CARBAMOIL FOSFATO DESHIDROGENASA (CPS)
- HIPERGLICEMIA NO CETÓSICA (NHK)
- ACIDURIA ARGININOSUCCÍNICA (ASL)
- CITRULINEMIA TIPO I (CIT-I)
- CITRULINEMIA TIPO II (CIT-II)
- TIROSINEMIA TIPO III (TYR-III)
- TIROSINEMIA TIPO II (TYR-II)
- TIROSINEMIA TIPO I (TYR-I)
- TIROSINEMIA TRANSITORIA NEONATAL (TYR-TRANS)
- ENFERMEDAD DE LA ORINA DEL JARABE DE ARCE CLÁSICA (MSUD)
- ENFERMEDAD CON ORINA OLOR A JARABE DE MAPLE INTERMEDIA
- HOMOCISTINURIA (HCY)
- HIPERMETIONINEMIA (MET)
- ATROFIA GIRATA
- DEFICIENCIA ORNITINA TRANSCARBAMILASA (OTC)
- HIPERORMITINEMIA-HIPERAMONEMIA-HOMOCITRULINURIA (HHH)
- FENILCETONURIA (PKU)
- HIPERFENILALANINEMIA (H-PHE)
- DEFECTO EN LA SÍNTESIS DEL COFACTOR DE BIOPTERINA (BIOP BS)
- DEFECTO DE REGENERACIÓN DEL COFACTOR BIOPTERINA (BIOP REG)
- PROLINEMIA
- ARGININEMIA (ARG)
- DEFICIENCIA DE BETA-CETOTIOLASA (BKT)
- 5-OXIPROLINURIA
- DEFICIENCIA DE CARBOXILASA PIRUVATO (PV)
- DEFICIENCIA TETRAHIDROFOLATO METILENO REDUCTASA (MTHFR)
- DEFICIENCIA DE TETRAHIDROBIOTERINA (BH4D)
- HIPERPROLINEMIA TIPO I (HP-I)
- HIPERPROLINEMIA TIPO II (HP-II)
- AMINOACIDURIA DICARBOXÍLICA
- DESÓRDENES DE DEFICIENCIA DE SERINA
- HIPERLYSINAEMIA, INTOLERANCIA PROTEINA LISINÚRICA (LPI)
- DESÓRDENES DEL CICLODE LA UREA (UCDs)
- VALINEMIA
- ACIDEMIA 3-HIDROXI-3-METILGLUTÁRICA (HMG)
- ISOBUTIRILGLICINURIA (IBG)
- ACIDEMIA ISOVALÉRICA (IVA)
- DEFICIENCIA DE 2-METILBUTIRIL-CoA DESHIDROGENASA (2MBG)
- DEFICIENCIA DE 3-METILCROTONIL-CoA CARBOXILASA (3MCC)
- ACIDEMIA 3-METILGLUTAXÓNICA (3MGA)
- ACIDEMIA 2-METIL-3-HIDROXIBUTÍRICA (2M3HBA)
- ACIDEMIA METILMALÓNICA (CBL A, CBL B)
- ACIDEMIA METILMALÓNICA Y HOMOCISTINURIA (CBL C, CBL D)
- DEFICIENCIA DE COBALAMINA (B12 DEF)
- DEFICIENCIA MITOCONDRIAL ACETOACETIL-CoA TIOLASA (BKT)
- ACIDEMIA PROPIÓNICA (PA)
- ACIDEMIA MALÓNICA (MAL)
- DEFICIENCIA MULTIPLE-CoA CARBOXILASA (MCD)
- ACIDEMIA GLUTARICA TIPO I (GA-I)
- ENCEFALOPATÍA ETILMALÓNICA (EE)
- ACIDEMIA FORMIMINOGLUTÁMICA (FIGLU)
- DEFICIENCIA 2,4-DIENOIL-CoA REDUCTASA (DE-RED)
- DEFICIENCIA DE METILENTETRAHIDROFOLATO REDUCTASA (MTHFR)
- ACIDEMIA METILMALÓNICA (MUT)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA MEDIA (MCAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA MUY LARGA (VLCAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA LARGA (LCHAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA CORTA (SCAD)
- DEFICIENCIA 3-HIDROXIACIL CoA DESHIDROGENASA CADENA CORTA/MEDIA (M/SCHADD)
- DEFICIENCIA DE CETOACIL CoA-TIOLASA DE CADENA MEDIA (MCKAT)
- DEFICIENCIA DEL TRASPORTADOR DE CARNITINA (CUD)
- DEFICIENCIA DE CARNITIN-ACILCARNTINA TRANSLOCASA (CACT)
- DEFICIENCIA DE LA CARNITIN PALMITOILTRANSFERASA I (CPT-I)
- DEFICIENCIA DE LA CARNITIN PALMITOILTRANSFERASA II (CPT-II)
- ACIDEMIA GLUTÁRICA TIPO II (GA-II)
- DEFICIENCIA DE LA PROTEÍNA TRIFUNCIONAL (TPF)
- DEFECTOS DE SÍNTESIS/INGESTA DE CARNITINA MATERNA
- DEFECTOS DE LA SÍNTESIS/INGESTA DE VITAMINA B12 MATERNA
- ACIDEMIA ETILMALÓNICA
Tamiz Metabólico Ampliado Plus -110 enfermedades
- HIPERPLASIA SUPRARENAL CONGÉNITA VIRALIZANTE SIMPLE
- HIPERPLASIA SUPRARENAL CONGÉNITA PERDEDORA DE SAL
- HIPOTIROIDISMO CONGÉNITO
- HIPERTIROTROPINEMIA
- DEFICIENCIA DE BIOTINIDASA
- FIBROSIS QUÍSTICA
- DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA
- GALACTOSEMIA TIPO DUARTE
- GALACTOSEMIA CLÁSICA
- GALACTOSEMIA TRANSITORIA
- DEFICIENCIA DE GALACTOQUINASA
- DEFICIENCIA DE GALACTOSA EPIMERASA
- ENFERMEDAD DE HEMOGLOBINA S
- ENFERMEDAD DE HEMOGLOBINA C
- ENFERMEDAD DE HEMOGLOBINA S/C
- ENFERMEDAD DE HEMOGLOBINA E
- ENFERMEDAD DE HEMOGLOBINA D
- ENFERMEDAD DE CÉLULAS FALCIFORMES CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA C CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA E CON BETA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA H
- ENFERMEDAD DE HEMOGLOBINA S CON RASGO DE ALFA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA S/C CON RASGO DE ALFA TALASEMIA
- ENFERMEDAD DE HEMOGLOBINA G FILADELFIA
- ENFERMEDAD DE HEMOGLOBINA G CON RASGO DE ALFA TALASEMIA
- BETA TALASEMIA MAYOR
- ANEMIA DREPANOCÍTICA
- DEFICIENCIA DE CARBAMOIL FOSFATO DESHIDROGENASA (CPS)
- HIPERGLICEMIA NO CETÓSICA (NHK)
- ACIDURIA ARGININOSUCCÍNICA (ASL)
- CITRULINEMIA TIPO I (CIT-I)
- CITRULINEMIA TIPO II (CIT-II)
- TIROSINEMIA TIPO III (TYR-III)
- TIROSINEMIA TIPO II (TYR-II)
- TIROSINEMIA TIPO I (TYR-I)
- TIROSINEMIA TRANSITORIA NEONATAL (TYR-TRANS)
- ENFERMEDAD DE LA ORINA DEL JARABE DE ARCE CLÁSICA (MSUD)
- ENFERMEDAD CON ORINA OLOR A JARABE DE MAPLE INTERMEDIA
- HOMOCISTINURIA (HCY)
- HIPERMETIONINEMIA (MET)
- ATROFIA GIRATA
- DEFICIENCIA ORNITINA TRANSCARBAMILASA (OTC)
- HIPERORMITINEMIA-HIPERAMONEMIA-HOMOCITRULINURIA (HHH)
- FENILCETONURIA (PKU)
- HIPERFENILALANINEMIA (H-PHE)
- DEFECTO EN LA SÍNTESIS DEL COFACTOR DE BIOPTERINA (BIOP BS)
- DEFECTO DE REGENERACIÓN DEL COFACTOR BIOPTERINA (BIOP REG)
- PROLINEMIA
- ARGININEMIA (ARG)
- DEFICIENCIA DE BETA-CETOTIOLASA (BKT)
- 5-OXIPROLINURIA
- DEFICIENCIA DE CARBOXILASA PIRUVATO (PV)
- DEFICIENCIA TETRAHIDROFOLATO METILENO REDUCTASA (MTHFR)
- DEFICIENCIA DE TETRAHIDROBIOTERINA (BH4D)
- HIPERPROLINEMIA TIPO I (HP-I)
- HIPERPROLINEMIA TIPO II (HP-II)
- AMINOACIDURIA DICARBOXÍLICA
- DESÓRDENES DE DEFICIENCIA DE SERINA
- HIPERLYSINAEMIA, INTOLERANCIA PROTEINA LISINURICA (LPI)
- DESÓRDENES DEL CICLODE LA UREA (UCDs)
- VALINEMIA
- ACIDEMIA 3-HIDROXI-3-METILGLUTÁRICA (HMG)
- ISOBUTIRILGLICINURIA (IBG)
- ACIDEMIA ISOVALÉRICA (IVA)
- DEFICIENCIA DE 2-METILBUTIRIL-CoA DESHIDROGENASA (2MBG)
- DEFICIENCIA DE 3-METILCROTONIL-CoA CARBOXILASA (3MCC)
- ACIDEMIA 3-METILGLUTAXÓNICA (3MGA)
- ACIDEMIA 2-METIL-3-HIDROXIBUTÍRICA (2M3HBA)
- ACIDEMIA METILMALÓNICA (CBL A, CBL B)
- ACIDEMIA METILMALÓNICA Y HOMOCISTINURIA (CBL C, CBL D)
- DEFICIENCIA DE COBALAMINA (B12 DEF)
- DEFICIENCIA MITOCONDRIAL ACETOACETIL-CoA TIOLASA (BKT)
- ACIDEMIA PROPIÓNICA (PA)
- ACIDEMIA MALÓNICA (MAL)
- DEFICIENCIA MULTIPLE-CoA CARBOXILASA (MCD)
- ACIDEMIA GLUTÁRICA TIPO I (GA-I)
- ENCEFALOPATÍA ETILMALÓNICA (EE)
- ACIDEMIA FORMIMINOGLUTÁMICA (FIGLU)
- DEFICIENCIA 2,4-DIENOIL-CoA REDUCTASA (DE-RED)
- DEFICIENCIA DE METILENTETRAHIDROFOLATO REDUCTASA (MTHFR)
- ACIDEMIA METILMALÓNICA (MUT)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA MEDIA (MCAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA MUY LARGA (VLCAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA LARGA (LCHAD)
- DEFICIENCIA DE ACIL-CoA DESHIDROGENASA DE CADENA CORTA (SCAD)
- DEFICIENCIA 3-HIDROXIACIL CoA DESHIDROGENASA CADENA CORTA/MEDIA (M/SCHADD)
- DEFICIENCIA DE CETOACIL CoA-TIOLASA DE CADENA MEDIA (MCKAT)
- DEFICIENCIA DEL TRASPORTADOR DE CARNITINA (CUD)
- DEFICIENCIA DE CARNITIN-ACILCARNTINA TRANSLOCASA (CACT)
- DEFICIENCIA DE LA CARNITIN PALMITOILTRANSFERASA I (CPT-I)
- DEFICIENCIA DE LA CARNITIN PALMITOILTRANSFERASA II (CPT-II)
- ACIDEMIA GLUTÁRICA TIPO II (GA-II)
- DEFICIENCIA DE LA PROTEÍNA TRIFUNCIONAL (TPF)
- DEFECTOS DE SÍNTESIS/INGESTA DE CARNITINA MATERNA
- DEFECTOS DE LA SÍNTESIS/INGESTA DE VITAMINA B12 MATERNA
- ACIDEMIA ETILMALÓNICA
- INMUNODEFICIENCIA SEVERA COMBINADA (SCID)
- ATROFIA MUSCULAR ESPINAL (SMA)
- FABRY
- POMPE
- KRABBE
- NIENMANN-PICK (A/B)
- GAUCHER
- MPS I
- MPS II,
- MPS IIIB,
- MPS IVA
- MPS VI
- MPS VII
- MPS X
¿Dudas? Comunícate con nosotros

Toma a domicilio
Con un costo adicional *Este puede variar según tu ubicación.
